ファイル形式¶
ファイル形式の概要¶
パラメータファイル¶
- mdp
実行パラメータ、gmx grompp および gmx convert-tpr の入力
- m2p
入力:gmx xpm2ps
構造ファイル¶
トポロジーファイル¶
実行する入力ファイル¶
- tpr
システムトポロジー、パラメータ、座標、および速度(バイナリ、ポータブル)
軌跡ファイル¶
エネルギーファイル¶
その他のファイル¶
ファイル形式の詳細¶
atp¶
ATPファイルには、原子の種類に関する一般的な情報(原子番号と、原子質量単位での質量など)が含まれています。
arn¶
`arn`ファイルを使用すると、分子を、従来の力場名から、IUPAC/PDBで定義された名前に変更できます。これにより、視覚化と識別が容易になります。
cpt¶
cptというファイル拡張子は、ポータブルチェックポイントファイルを示します。シミュレーションの完全な状態がチェックポイントファイルに保存されており、これには拡張された熱力学/圧力平衡変数、乱数状態、およびNMRの時間平均データが含まれます。ドメイン分割を使用する場合、分割設定に関する情報も保存されます。
こちらも参照してください:gmx mdrun。
dat¶
ファイル拡張子が「.dat」であるファイルには、汎用的な入力または出力データが含まれています。すべてのデータファイル形式を分類することは不可能であるため、|Gromacs|には形式が定義されていない汎用ファイル形式「dat」が存在します。
edi¶
「edi」というファイル拡張を持つファイルには、Essential Dynamicsの制約を使用して分子動力学を実行するための「gmx mdrun」に必要な情報が含まれています。以前は、「WHAT IF」<https://swift.cmbi.umcn.nl/whatif/>プログラムのオプションを使用して、これらのファイルを生成することができました。
edr¶
「edr」というファイル拡張子は、ポータブルなエネルギーファイルを示します。エネルギーデータは、xdrプロトコルを使用して保存されます。
こちらも参照してください:gmx エネルギー。
ene¶
.eneファイル拡張子は、バイナリ形式のエネルギーファイルを示します。これは、`gmx mdrun`によって生成されたエネルギー値を保持します。
このファイルは、`gmx eneconv`プログラムを使用して、ハードウェアプラットフォーム間での互換性のある汎用ファイル(`edr`ファイル)に変換できます。
こちらも参照してください:gmx エネルギー。
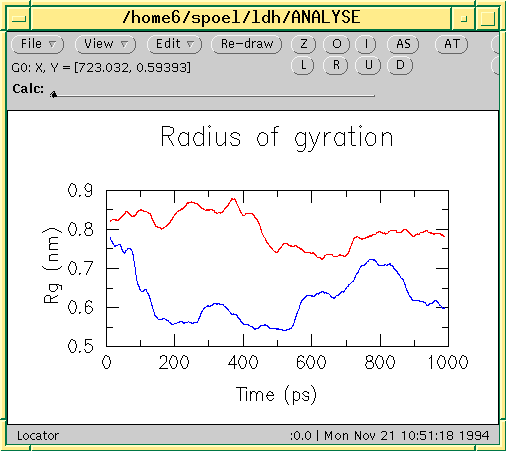
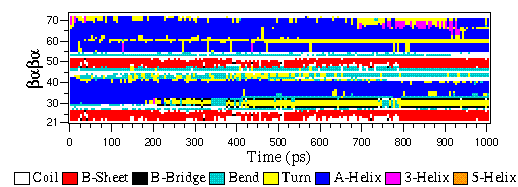
eps¶
EPSファイル形式は、特別な|Gromacs|形式ではなく、単に標準のPostScript(tm)のバリエーションです。以下に、:ref:`gmx xpm2ps`プログラムによって生成されたEPSファイルのサンプルを示します。これは、ペプチドの二次構造を時間とともに変化する様子を示しています。
g96¶
g96拡張子のファイルは、GROMOS-96の初期/最終設定ファイル、座標軌跡ファイル、または両方の組み合わせである可能性があります。ファイルは固定形式で、すべての浮動小数点数は15.9で記述されます(ファイルサイズが大きくなる可能性があります)。|Gromacs|は、以下のデータブロックを次の順序でサポートしています。
Header block:
タイトル(必須)フレームブロック:
TIMESTEP(オプション)
POSITION/POSITIONRED(必須)
VELOCITY/VELOCITYRED(オプション)
BOX(オプション)
完全な説明については、GROMOS-96のマニュアルを参照してください。
注意:|Gromacs|のすべてのプログラムは、圧縮されたファイルまたはg-zippedファイルを読み取ることができます。
gro¶
gro 拡張子のファイルには、Gromos87形式の分子構造が含まれています。groファイルは、ファイルを単純に連結することで、軌跡として使用できます。プログラムは、各フレームのタイトル文字列から時間値を読み取ろうとしますが、これは「t=」という形式で記述されている必要があります(例:以下を参照)。
以下にサンプルが含まれています:
MD of 2 waters, t= 0.0
6
1WATER OW1 1 0.126 1.624 1.679 0.1227 -0.0580 0.0434
1WATER HW2 2 0.190 1.661 1.747 0.8085 0.3191 -0.7791
1WATER HW3 3 0.177 1.568 1.613 -0.9045 -2.6469 1.3180
2WATER OW1 4 1.275 0.053 0.622 0.2519 0.3140 -0.1734
2WATER HW2 5 1.337 0.002 0.680 -1.0641 -1.1349 0.0257
2WATER HW3 6 1.326 0.120 0.568 1.9427 -0.8216 -0.0244
1.82060 1.82060 1.82060
行には以下の情報(上から下へ)が含まれています。
タイトル (自由形式の文字列、オプションで「t=」の後にPS単位での時間)
原子数 (自由形式の整数)
1行で1つの原子(固定形式、下記参照)を指定
ボックスベクトルの(自由形式、スペースで区切られた実数)、値:v1(x) v2(y) v3(z) v1(y) v1(z) v2(x) v2(z) v3(x) v3(y)。最後の6つの値は省略できます(0に設定されます)。|Gromacs|は、v1(y)=v1(z)=v2(z)=0のボックスのみをサポートします。
この形式は固定されており、つまりすべての列が固定された位置にあります。オプション(現時点ではtrjconvのみで)として、任意の桁数の小数を持つgroファイルを記述できます。その場合、形式は「n+5」の位置に「n」桁の小数(速度の場合は「n+1」桁)となり、「8」と「3」の代わりに、速度の場合は「4」桁になります。読み込む際には、小数点間の距離から精度が推論されます(これは「n+5」になります)。列には、左から右に向かって、以下の情報が含まれています:
残余番号 (5桁、整数)
残余名 (5 文字)
アトム名 (5文字)
アトム番号 (5桁、整数)
位置(単位:ナノメートル、3列に並べられたx、y、z座標。各座標は8桁で、小数点以下3桁)
(速度 (nm/ps (または km/s)、x y z を 3 列に配置、各列に 8 桁、4 桁の小数点))
注意: 個別の分子またはイオン(例:水またはCl-)は、残基として扱われます。|Gromacs|ライブラリを使用せずに、独自のプログラムでこれらのファイルを書き込む場合は、以下の形式を使用できます。
- C形式
"%5d%-5s%5s%5d%8.3f%8.3f%8.3f%8.4f%8.4f%8.4f"- フォーラン形式
(i5,2a5,i5,3f8.3,3f8.4)- 形式: パスカル
これはユーザーに任せてください。
注意: これは書き込み形式であり、上記の例に示すように、フィールドはスペースなしで記述できます。したがって、C言語で同じ形式の記述では読み取ることができません。
hdb¶
「hdb」というファイル拡張子は、水素データベースを示します。このファイルは、gmx pdb2gmx が、元のデータベースに存在しない水素原子、または -ignh オプションで削除された水素原子を構築するために必要です。
itp¶
itp`ファイル拡張子は、インクルード・トポロジーを意味します。これらのファイルは、トポロジーファイル(:ref:``top`拡張子を持つファイル)に含まれています。
ログ¶
ログファイルは、一部の|Gromacs|プログラムによって生成され、通常は人間が読める形式で保存されます。「more logfile」コマンドを使用します。
m2p¶
m2pファイル形式には、:ref:`gmx xpm2ps`プログラムの入力オプションが含まれています。これらのオプションは、:ref:`gmx xpm2ps`から生成されるPostScript(tm)出力を確認することで、非常に理解しやすいものです。
; Command line options of xpm2ps override the parameters in this file
black&white = no ; Obsolete
titlefont = Times-Roman ; A PostScript Font
titlefontsize = 20 ; Font size (pt)
legend = yes ; Show the legend
legendfont = Times-Roman ; A PostScript Font
legendlabel = ; Used when there is none in the .xpm
legend2label = ; Used when merging two xpm's
legendfontsize = 14 ; Font size (pt)
xbox = 2.0 ; x-size of a matrix element
ybox = 2.0 ; y-size of a matrix element
matrixspacing = 20.0 ; Space between 2 matrices
xoffset = 0.0 ; Between matrix and bounding box
yoffset = 0.0 ; Between matrix and bounding box
x-major = 20 ; Major ticks on x axis every .. frames
x-minor = 5 ; Id. Minor ticks
x-firstmajor = 0 ; First frame for major tick
x-majorat0 = no ; Major tick at first frame
x-majorticklen = 8.0 ; x-majorticklength
x-minorticklen = 4.0 ; x-minorticklength
x-label = ; Used when there is none in the .xpm
x-fontsize = 16 ; Font size (pt)
x-font = Times-Roman ; A PostScript Font
x-tickfontsize = 10 ; Font size (pt)
x-tickfont = Helvetica ; A PostScript Font
y-major = 20
y-minor = 5
y-firstmajor = 0
y-majorat0 = no
y-majorticklen = 8.0
y-minorticklen = 4.0
y-label =
y-fontsize = 16
y-font = Times-Roman
y-tickfontsize = 10
y-tickfont = Helvetica
mdp¶
詳細なオプションの説明については、ユーザーマニュアルを参照してください。
以下はサンプルMDPファイルです。項目の順序は重要ではありませんが、同じものを2回入力すると、**最後の**ものが使用されます(`gmx grompp`を使用すると、値を上書きした場合に注意喚起が表示されます)。左側のハイフンとアンダースコアは無視されます。
これらのオプションの値は、タンパク質が水溶液中で1ナノ秒間のシミュレーションを行う場合の値を表しています。
注記: 使用する力場に応じて選択されたパラメータ(例: 短距離カットオフ)は異なります。
integrator = md
dt = 0.002
nsteps = 500000
nstlog = 5000
nstenergy = 5000
nstxout-compressed = 5000
continuation = yes
constraints = all-bonds
constraint-algorithm = lincs
cutoff-scheme = Verlet
coulombtype = PME
rcoulomb = 1.0
vdwtype = Cut-off
rvdw = 1.0
DispCorr = EnerPres
tcoupl = V-rescale
tc-grps = Protein SOL
tau-t = 0.1 0.1
ref-t = 300 300
pcoupl = Parrinello-Rahman
tau-p = 2.0
compressibility = 4.5e-5
ref-p = 1.0
この入力により、ref:gmx grompp は、デフォルトのファイル名 mdout.mdp でコメント付きのファイルを作成します。そのファイルには、上記のオプションに加えて、明示的に設定されていないすべてのオプションとそのデフォルト値が含まれます。
上記のmdpサンプルは、mdpeditor を使用して生成されました。このツールを使用すると、簡単な指示に基づいて、mdpファイルの作成をベストプラクティスに従って行うことができます。
mtx¶
ファイルには「mtx」という拡張子がついており、行列が含まれています。ファイル形式は:ref:`trr`形式と同じです。現在、このファイル形式は、:ref:`gmx mdrun`で生成され、:ref:`gmx nmeig`で読み込まれるヘッセ行列のみで使用されています。
ndx¶
GROMACS のインデックスファイル(通常は index.ndx と呼ばれます)には、ユーザーが定義できる原子のセットが含まれています。このファイルは、ほとんどの分析プログラムと、プレプロセッサ(gmx grompp)で読み取ることができます。これらのプログラムは、インデックスファイルが提供されていない場合、デフォルトのインデックスグループを作成するため、特別なグループが必要な場合にのみインデックスファイルを作成する必要があります。
まず、グループ名は角括弧で囲みます。 以下のアトム番号は、好きなだけ複数行にわたって記述できます。 アトムの番号は1から始まります。
例となるファイルはここにあります:
[ Oxygen ]
1 4 7
[ Hydrogen ]
2 3 5 6
8 9
存在するのは2つのグループで、合計9つの原子があります。最初のグループである**酸素**には3つの元素が含まれています。2番目のグループである**水素**には6つの元素が含まれています。
インデックスファイルの生成ツールが利用可能です: gmx make_ndx。
n2t¶
この GROMACS ファイルを使用すると、構造ファイルに存在する原子名の間の基本的な変換を実行できます。これは、主に gmx x2top などのユーティリティを使用する際に役立ちますが、このファイルに記載されている情報は非常に限られていることに注意してください。
例となるファイル(share/top/gromos53a5.ff/atomname2type.n2t)はここにあります:
H H 0.408 1.008 1 O 0.1
O OA -0.674 15.9994 2 C 0.14 H 0.1
C CH3 0.000 15.035 1 C 0.15
C CH0 0.266 12.011 4 C 0.15 C 0.15 C 0.15 O 0.14
ファイル形式の簡単な説明は以下のとおりです:
列 1: 原子/原子名の最初の文字。
列 2: 割り当てるべきアトームのタイプ。
3列目: 割り当てるべき料金。
4列目: 粒子の質量。
列 5: この原子が結合されている他の原子の数 N。この数に関連するフィールドが次のフィールドです。各原子について、元素記号と結合長の参照距離が記載されています。
6列以降:N接続(5列に記載されている)の原子への参照結合長と、パラメータが割り当てられている原子(1列)の元素記号。参照結合長は、このファイルに指定された値から±10%の範囲内である必要があります。この範囲外の結合は、パラメータが割り当てられている原子に接続されていると認識されません。
出力¶
ファイルには「out」という拡張子が付いている場合、一般的な出力データが含まれています。すべてのデータファイル形式を分類することは不可能であるため、|Gromacs|には「out」という汎用的なファイル形式があり、具体的な形式は定義されていません。
pdb¶
ファイル拡張子が pdb のファイルは、タンパク質データベースのファイル形式における分子構造ファイルです。タンパク質データベースのファイル形式は、分子構造における原子の位置を記述します。座標は、ATOM および HETATM レコードから読み取られ、ファイルが終了するか、ENDMDL レコードが遭遇するまで続きます。GROMACS プログラムは、CRYST1 エントリ内のシミュレーションボックスを読み書きできます。また、pdb 形式は、複数の構造(ENDMDL で区切られたもの)を 1 つのファイルから読み取ったり、書き込んだりするためのトレース形式としても使用できます。
例¶
PDBファイルは、以下のようになります:
ATOM 1 H1 LYS 1 14.260 6.590 34.480 1.00 0.00
ATOM 2 H2 LYS 1 13.760 5.000 34.340 1.00 0.00
ATOM 3 N LYS 1 14.090 5.850 33.800 1.00 0.00
ATOM 4 H3 LYS 1 14.920 5.560 33.270 1.00 0.00
...
...
rtp¶
「rtp」というファイル拡張子は、残差トポロジーを意味します。このようなファイルは、:ref:`gmx pdb2gmx`を使用して、:ref:`pdb`ファイルに存在するタンパク質の|Gromacs|トポロジーを作成するために必要です。このファイルには、4つの結合相互作用と残差に関するデフォルトの相互作用タイプと、原子とオプションで結合、角度、二面体、および不適切な要素で構成される残差エントリが含まれています。結合、角度、二面体、および不適切な要素にパラメータを追加できます。これらのパラメータは、:ref:`itp`ファイル内の標準パラメータを上書きします。これは、特別な場合にのみ使用する必要があります。パラメータの代わりに、各結合相互作用に対して文字列を追加できます。この文字列は:ref:`top`ファイルにコピーされ、GROMOS96力場で使用されます。
gmx pdb2gmx はすべての角度を自動的に生成するため、[angles] フィールドは主に itp パラメータを上書きするためにのみ使用されます。
gmx pdb2gmx は、回転可能な結合ごとに適切なジヘドラルを自動的に生成します。理想的には、重い原子に配置されます。 [dihedrals] フィールドを使用すると、指定されたジヘドラルに対応する結合には、他のジヘドラルは生成されません。 1 つの回転可能な結合に複数のジヘドラルを配置することも可能です。
gmx pdb2gmx は、除外する数を3に設定します。これは、最大3つの結合でつながっている原子間の相互作用を除外することを意味します。すべての原子ペア(水素を除く)のうち、3つの結合で隔てられているペアに対して、相互作用が生成されます。さらに多くの相互作用を除外する必要がある場合、または特定の原子ペアの相互作用を生成しないようにするには、[exclusions] フィールドを追加し、その後に原子名のペアを別々の行に記述します。これらの原子間のすべての非結合およびペアの相互作用が除外されます。
以下にサンプルを示します。
[ bondedtypes ] ; mandatory
; bonds angles dihedrals impropers
1 1 1 2 ; mandatory
[ GLY ] ; mandatory
[ atoms ] ; mandatory
; name type charge chargegroup
N N -0.280 0
H H 0.280 0
CA CH2 0.000 1
C C 0.380 2
O O -0.380 2
[ bonds ] ; optional
;atom1 atom2 b0 kb
N H
N CA
CA C
C O
-C N
[ exclusions ] ; optional
;atom1 atom2
[ angles ] ; optional
;atom1 atom2 atom3 th0 cth
[ dihedrals ] ; optional
;atom1 atom2 atom3 atom4 phi0 cp mult
[ impropers ] ; optional
;atom1 atom2 atom3 atom4 q0 cq
N -C CA H
-C -CA N -O
[ ZN ]
[ atoms ]
ZN ZN 2.000 0
r2b¶
`r2b`ファイルは、異なる力場において異なる名前を持つ残基、またはプロトン化状態に応じて異なる名前を持つ残基の名前を変換します。
tdb¶
`tdb`ファイルには、ポリペプチド鎖の末端に配置できるアミノ酸に関する情報が含まれています。
tex¶
私達は LaTeX を ドキュメント の処理に使用しています。入力はユーザーにとって使いにくいですが、ワープロソフトにはないいくつかの利点があります。
LaTeX は、フォーマットに関する知識が豊富で、おそらくあなたよりも多い。
入力は明確で、常に自分が何をしようとしているのかわかります。
あらゆるもの、手紙から論文まで作成できます
もっと詳しく...
tng¶
.tng というファイル拡張を持つファイルには、シミュレーションの軌跡に関連するさまざまなデータが含まれる可能性があります。たとえば、座標、速度、力、および/またはエネルギーが含まれている場合があります。さまざまな :ref:`mdp ファイルオプションは、これらのうちどれが :ref:`gmx mdrun によって書き込まれるか、データが圧縮された状態で書き込まれるか、および圧縮の度合いを制御します。このファイルは、ポータブルなバイナリ形式で保存されており、:ref:`gmx dump を使用して読み取ることができます。
gmx ダンプ -f traj.tng
または、もしあなたが速く読まない場合は:
gmx dump -f traj.tng | less
また、ファイルのコンテンツ(フレーム数など)を簡単に確認することもできます。
gmx チェック -f traj.tng
トップ¶
最上位のファイル拡張子は、トポロジーを示します。これは、`gmx grompp`によって読み込まれるASCIIファイルであり、このファイルが処理され、バイナリ形式のトポロジーファイル(:ref:`tpr`ファイル)が作成されます。
以下にサンプルファイルが含まれています:
;
; Example topology file
;
[ defaults ]
; nbfunc comb-rule gen-pairs fudgeLJ fudgeQQ
1 1 no 1.0 1.0
; The force field files to be included
#include "rt41c5.itp"
[ moleculetype ]
; name nrexcl
Urea 3
[ atoms ]
; nr type resnr residu atom cgnr charge
1 C 1 UREA C1 1 0.683
2 O 1 UREA O2 1 -0.683
3 NT 1 UREA N3 2 -0.622
4 H 1 UREA H4 2 0.346
5 H 1 UREA H5 2 0.276
6 NT 1 UREA N6 3 -0.622
7 H 1 UREA H7 3 0.346
8 H 1 UREA H8 3 0.276
[ bonds ]
; ai aj funct c0 c1
3 4 1 1.000000e-01 3.744680e+05
3 5 1 1.000000e-01 3.744680e+05
6 7 1 1.000000e-01 3.744680e+05
6 8 1 1.000000e-01 3.744680e+05
1 2 1 1.230000e-01 5.020800e+05
1 3 1 1.330000e-01 3.765600e+05
1 6 1 1.330000e-01 3.765600e+05
[ pairs ]
; ai aj funct c0 c1
2 4 1 0.000000e+00 0.000000e+00
2 5 1 0.000000e+00 0.000000e+00
2 7 1 0.000000e+00 0.000000e+00
2 8 1 0.000000e+00 0.000000e+00
3 7 1 0.000000e+00 0.000000e+00
3 8 1 0.000000e+00 0.000000e+00
4 6 1 0.000000e+00 0.000000e+00
5 6 1 0.000000e+00 0.000000e+00
[ angles ]
; ai aj ak funct c0 c1
1 3 4 1 1.200000e+02 2.928800e+02
1 3 5 1 1.200000e+02 2.928800e+02
4 3 5 1 1.200000e+02 3.347200e+02
1 6 7 1 1.200000e+02 2.928800e+02
1 6 8 1 1.200000e+02 2.928800e+02
7 6 8 1 1.200000e+02 3.347200e+02
2 1 3 1 1.215000e+02 5.020800e+02
2 1 6 1 1.215000e+02 5.020800e+02
3 1 6 1 1.170000e+02 5.020800e+02
[ dihedrals ]
; ai aj ak al funct c0 c1 c2
2 1 3 4 1 1.800000e+02 3.347200e+01 2.000000e+00
6 1 3 4 1 1.800000e+02 3.347200e+01 2.000000e+00
2 1 3 5 1 1.800000e+02 3.347200e+01 2.000000e+00
6 1 3 5 1 1.800000e+02 3.347200e+01 2.000000e+00
2 1 6 7 1 1.800000e+02 3.347200e+01 2.000000e+00
3 1 6 7 1 1.800000e+02 3.347200e+01 2.000000e+00
2 1 6 8 1 1.800000e+02 3.347200e+01 2.000000e+00
3 1 6 8 1 1.800000e+02 3.347200e+01 2.000000e+00
[ dihedrals ]
; ai aj ak al funct c0 c1
3 4 5 1 2 0.000000e+00 1.673600e+02
6 7 8 1 2 0.000000e+00 1.673600e+02
1 3 6 2 2 0.000000e+00 1.673600e+02
; Include SPC water topology
#include "spc.itp"
[ system ]
Urea in Water
[ molecules ]
Urea 1
SOL 1000
tpr¶
`tpr`ファイル拡張子は、ポータブルバイナリ実行入力ファイルを示します。このファイルには、シミュレーションの開始構造、分子のトポロジー、およびすべてのシミュレーションパラメータが含まれています。このファイルはバイナリ形式であるため、通常のテキストエディタでは開くことができません。ポータブルバイナリ実行入力ファイルを読み込むには、次のコマンドを入力します。
gmx ダンプ -s topol.tpr
または、もしあなたが速く読まない場合は:
gmx dump -s topol.tpr | less
また、以下のコマンドを使用して2つの tpr ファイルを比較することもできます。
gmx チェック -s1 top1 -s2 top2 | less
trr¶
ファイル拡張子が.trrであるファイルには、シミュレーションの軌跡が含まれています。このファイルには、|Gromacs|にmdpファイルで指定したように、すべての座標、速度、力、およびエネルギーが記録されています。このファイルは、ポータブルなバイナリ形式で、:ref:`gmx dump`で読み取ることができます。
gmx dump -f traj.trr
または、もしあなたが速く読まない場合は:
gmx dump -f traj.trr | less
また、ファイルのコンテンツ(フレーム数など)を簡単に確認することもできます。
% gmx チェック -f traj.trr
vsd¶
VSDファイルには、特定の力場内で複数の分子上に仮想サイトを配置する方法に関する情報が含まれています。
xdr¶
GROMACS は、内部的に座標ファイルなどの情報を保存するために、XDRファイル形式を使用します。
xpm¶
GROMACS の xpm ファイル形式は、XPixMap 形式と互換性があり、行列データを保存するために使用されます。したがって、GROMACS の xpm ファイルは、XV などのプログラムで直接表示できます。また、GIMP にインポートして、フォントとグラフィックスに対して強いアンチエイリアシングを使用して、300 DPI に拡大することも可能です。xpm ファイル内の最初の行列データ行は、最後の行列行に対応します。XPixMap 形式に加えて、GROMACS の xpm ファイルには、追加のフィールドが含まれる場合があります。これらのフィールドに記載されている情報は、gmx xpm2ps を使用して xpm ファイルを EPS 形式に変換する際に使用されます。追加のフィールドは次のとおりです。
``gv_xpm``宣言の前に、「title」、「legend」、「x-label」、「y-label」および「type」という文字列が続きます。 「legend」フィールドは凡例のタイトルを決定します。「type」フィールドには、「continuous」または「discrete」という文字列が続く必要があり、これはEPSファイルで描画される凡例の種類を決定します。デフォルトのタイプは「continuous」です。
XPM カラーマップのエントリには、文字列が続く場合があります。これは、その色に付けるラベルです。
カラーマップと行列データの間には、「x-軸」および/または「y-軸」のフィールドと、それに対応する軸の目盛りが存在することがあります。
以下に示す GROMACS xpm ファイルには、すべての追加フィールドが含まれています。 Cコメントの区切り文字と、追加フィールド内のコロンはオプションです。
/* XPM */
/* This matrix is generated by gmx rms. */
/* title: "Backbone RMSD matrix" */
/* legend: "RMSD (nm)" */
/* x-label: "Time (ps)" */
/* y-label: "Time (ps)" */
/* type: "Continuous" */
static char * gv_xpm[] = {
"13 13 6 1",
"A c #FFFFFF " /* "0" */,
"B c #CCCCCC " /* "0.0399" */,
"C c #999999 " /* "0.0798" */,
"D c #666666 " /* "0.12" */,
"E c #333333 " /* "0.16" */,
"F c #000000 " /* "0.2" */,
/* x-axis: 0 40 80 120 160 200 240 280 320 360 400 440 480 */
/* y-axis: 0 40 80 120 160 200 240 280 320 360 400 440 480 */
"FEDDDDCCCCCBA",
"FEDDDCCCCBBAB",
"FEDDDCCCCBABC",
"FDDDDCCCCABBC",
"EDDCCCCBACCCC",
"EDCCCCBABCCCC",
"EDCCCBABCCCCC",
"EDCCBABCCCCCD",
"EDCCABCCCDDDD",
"ECCACCCCCDDDD",
"ECACCCCCDDDDD",
"DACCDDDDDDEEE",
"ADEEEEEEEFFFF"
xtc¶
`xtc`形式は、軌跡データの**互換性のある**形式です。これは、Unix NFSシステム向けに作成された*xdr*ルーチンを使用してデータを書き込みおよび読み込みます。軌跡は、以下の手順で動作する、低精度アルゴリズムを使用して書き込まれます。まず、(nm単位)の座標に、通常は1000のスケールファクターを掛けて、pm単位の座標を得ます。次に、これらの値を整数に丸めます。その後、いくつかのテクニックが適用されます。例えば、隣接する原子は通常、空間的に近い(例えば、水分子)という事実を利用します。そのため、3D浮動小数点座標を書き込むための特別なルーチンを備えた*xdr*ライブラリが拡張されます。このルーチンは、当初、Frans van HoeselがEuroportプロジェクトの一部として作成しました。最新版は、`このリンク <https://github.com/Pappulab/xdrf>`から入手できます。
すべてのデータは、xdr ユーティリティへの呼び出しを使用して保存されます。
- int magic
現在のファイルバージョンに対応するマジックナンバーは1995です。
- int natoms
軌跡中の原子の数。
- int step
シミュレーションステップ。
- 浮動小数点数 時間
シミュレーション時間。
- float box[3][3]
計算箱は、トリキリン形のPBCを可能にする3つの基底ベクトルの集合として保存されます。直方体の場合、箱の辺は行列の対角線上にあるように保存されます。
- 3dfcoord x[natoms]
座標データは、低精度で保存されます。ただし、原子数が9未満の場合、低精度は使用されません。
C++プログラムでのxtcの使用¶
以下のような圧縮された .xtc 形式のファイルを利用するために、独自の解析ツールを作成することも可能です。例として、インストールディレクトリ内の share/gromacs/template ディレクトリにある template.cpp ファイルを参照してください。また、詳細については、https://manual.gromacs.org/current/doxygen/html-full/page_analysistemplate.xhtml を参照してください。
以下のルーチンは、``xtcio.h``を通じて、xtcファイルの読み書きを行うために利用できます。
/* All functions return 1 if successful, 0 otherwise */
struct t_fileio* open_xtc(const char* filename, const char* mode);
/* Open a file for xdr I/O */
void close_xtc(struct t_fileio* fio);
/* Close the file for xdr I/O */
int read_first_xtc(struct t_fileio* fio,
int* natoms,
int64_t* step,
real* time,
matrix box,
rvec** x,
real* prec,
gmx_bool* bOK);
/* Open xtc file, read xtc file first time, allocate memory for x */
int read_next_xtc(struct t_fileio* fio, int natoms, int64_t* step, real* time, matrix box, rvec* x, real* prec, gmx_bool* bOK);
/* Read subsequent frames */
int write_xtc(struct t_fileio* fio, int natoms, int64_t step, real time, const rvec* box, const rvec* x, real prec);
/* Write a frame to xtc file */
ライブラリ関数を使用するには、ファイルに "gromacs/fileio/xtcio.h" を含め、 -lgromacs でリンクしてください。
xvg¶
GROMACS の分析ツールからのほとんどの出力は、Grace(以前はXmgrと呼ばれていたもの)への入力として利用できます。Graceは非常に柔軟で、またオープンソースであるため、当社で使用しています。PostScript(tm)形式の出力を生成し、例えばLaTeXドキュメントやその他のワープロソフトへの組み込みに適しています。
以下に、|Gromacs|データを含むGraceセッションの一例を示します。